Paired-seq#
Check this GitHub page to see how Paired-seq libraries are generated experimentally. This is a split-pool based combinatorial indexing method, where open chromatin DNA are transposed by indexed homo-dimer Tn5 and mRNA molecules are reverse transcribed by barcoded oligo-dT primers. Then three rounds of ligation are performed to added three 7-bp barcodes. UMI is added at the last round of ligation. After that, the reaction was split into two portion, one for ATAC and the other for RNA library preparations. Single cells can be identified by the combination of the Tn5/RT barcode plus three 7-bp barcodes.
For Your Own Experiments#
If you follow the protocol from the paper, you should have two libraries per sample: one for RNA and the other for ATAC. The libraries consists of standard Illumina adapters (Nextera + TruSeq), so you could sequence them via your core facility or a company or by yourself. If you sequence your data via your core facility or a company, you will need to provide the sample and modality index sequence. In this case, it is basically i7 + i5. They will demultiplex for you and give you back two fastq files per sample per modality.
Your sequencing read configuration is like this:
Order |
Read |
Cycle |
Description |
|---|---|---|---|
1 |
Read 1 |
>50 |
This yields |
2 |
Index 1 (i7) |
8 or 10 |
This yields |
3 |
Index 2 (i5) |
8 or 10 |
This yields |
4 |
Read 2 |
130 |
This yields |
The 4th read (Read 2) has the following information, which will be used to identify single cells (NOTE: LB means Ligation Barcode, see later section about whitelist, then you will understand):
Modality |
Length |
Sequence (5’ -> 3’) |
|---|---|---|
ATAC |
130 bp |
10 bp UMI + 7 bp |
RNA |
130 bp |
10 bp UMI + 7 bp |
The 0 - 3 random bases (N) in the middle is there to increase the base complexities.
If you sequence by yourself, you need to run bcl2fastq by yourself with a SampleSheet.csv and without the --create-fastq-for-index-reads flag, because they are not really needed after demultiplexing. Here is an example of SampleSheet.csv of a NextSeq run with two different samples using the standard indexing primers from the Paired-seq paper (6-bp i7, 8-bp i5):
[Header],,,,,,,,,,,
IEMFileVersion,5,,,,,,,,,,
Date,17/12/2019,,,,,,,,,,
Workflow,GenerateFASTQ,,,,,,,,,,
Application,NextSeq FASTQ Only,,,,,,,,,,
Instrument Type,NextSeq/MiniSeq,,,,,,,,,,
Assay,AmpliSeq Library PLUS for Illumina,,,,,,,,,,
Index Adapters,AmpliSeq CD Indexes (384),,,,,,,,,,
Chemistry,Amplicon,,,,,,,,,,
,,,,,,,,,,,
[Reads],,,,,,,,,,,
53,,,,,,,,,,,
130,,,,,,,,,,,
,,,,,,,,,,,
[Settings],,,,,,,,,,,
,,,,,,,,,,,
[Data],,,,,,,,,,,
Sample_ID,Sample_Name,Sample_Plate,Sample_Well,Index_Plate,Index_Plate_Well,I7_Index_ID,index,I5_Index_ID,index2,Sample_Project,Description
Sample01_ATAC,,,,,,P701,ATCACG,N502,ATAGAGAG,,
Sample01_RNA,,,,,,P702,CGATGT,N503,AGAGGATA,,
Sample02_ATAC,,,,,,P703,TTAGGC,N504,TCTACTCT,,
Sample02_RNA,,,,,,P704,TGACCA,N505,CTCCTTAC,,
Simply run bcl2fastq like this:
bcl2fastq --no-lane-splitting \
--ignore-missing-positions \
--ignore-missing-controls \
--ignore-missing-filter \
--ignore-missing-bcls \
-r 4 -w 4 -p 4
After this, you will have R1_001.fastq.gz and R2_001.fastq.gz per sample per modality. Using the examples above, these are the files you should get:
# Sample 01 ATAC
Sample01_ATAC_S1_R1_001.fastq.gz # 50 bp: genomic insert
Sample01_ATAC_S1_R2_001.fastq.gz # 130 bp: UMI and Cell barcodes
# Sample 01 RNA
Sample01_RNA_S2_R1_001.fastq.gz # 50 bp: cDNA insert
Sample01_RNA_S2_R2_001.fastq.gz # 130 bp: UMI and Cell barcodes
# Sample 02 ATAC
Sample02_ATAC_S3_R1_001.fastq.gz # 50 bp: genomic insert
Sample02_ATAC_S3_R2_001.fastq.gz # 130 bp: UMI and Cell barcodes
# Sample 02 RNA
Sample02_RNA_S4_R1_001.fastq.gz # 50 bp: cDNA insert
Sample02_RNA_S4_R2_001.fastq.gz # 130 bp: UMI and Cell barcodes
You are ready to go from here.
Public Data#
We will use the data from the following paper:
Note
Zhu C, Yu M, Huang H, Juric I, Abnousi A, Hu R, Lucero J, Behrens MM, Hu M, Ren B (2019) An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat Struct Mol Biol 26:1063–1070. https://doi.org/10.1038/s41594-019-0323-x
where the authors developed Paired-seq for the first time and described the technical details of the method. The data is deposited to GEO under the accession code GSE130399. There are quite a few samples, and we are going to use the Adult Cerebral Cortex Lib#1 sample (SRR8980190 for ATAC, and SRR8980191 for RNA). We could simply download the fastq files from ENA:
mkdir -p paired-seq/data
wget -P paired-seq/data -c \
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR898/000/SRR8980190/SRR8980190_1.fastq.gz \
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR898/000/SRR8980190/SRR8980190_2.fastq.gz \
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR898/001/SRR8980191/SRR8980191_1.fastq.gz \
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR898/001/SRR8980191/SRR8980191_2.fastq.gz
According to the Paired-seq publication, Read 1 should be 53 bp and Read 2 should be 130 bp. However, if you look inside the downloaded files, you will see reads with variable lengths with a maximum 53 bp in Read 1 and 130 bp in Read 2. Not sure what happened, but let’s remove the shorter reads. In this case, you need Cutadapt or the like. The commands are:
# Clean ATAC
cutadapt -j 4 -m 53:130 -o paired-seq/data/SRR8980190_cleaned_R1.fastq.gz -p paired-seq/data/SRR8980190_cleaned_R2.fastq.gz paired-seq/data/SRR8980190_1.fastq.gz paired-seq/data/SRR8980190_2.fastq.gz
# Clean RNA
cutadapt -j 4 -m 53:130 -o paired-seq/data/SRR8980191_cleaned_R1.fastq.gz -p paired-seq/data/SRR8980191_cleaned_R2.fastq.gz paired-seq/data/SRR8980191_1.fastq.gz paired-seq/data/SRR8980191_2.fastq.gz
The -m v1[:v2] option will remove Read 1 sequence with less than v1 in length and Read 2 sequence with less than v2 in length. Now the cleaned_R1.fastq.gz and cleaned_R2.fastq.gz is our starting point.
Now we are ready to process the RNA data. However, for ATAC data, we need some extra work, because the 0 - 3 random bases (N) in the middle of Read 2 make the situation more complicated. The random bases have different length: None, 1, 2 or 3. This makes the barcode position variable in different reads, and chromap currently does not support such complex barcodes. Therefore, we need to extract the cell barcodes with UMI-tools, because it supports regular expression. This step is kind of slow, especially for large data sets:
umi_tools extract --extract-method=regex \
--bc-pattern2='(?P<umi_1>.{10})(?P<cell_1>.{7}).{0,3}(?P<discard_1>GTGGCCGATGTTTCGCATCGGCGTACGACT){s<=3}(?P<cell_2>.{7})(?P<discard_2>GGATTCGAGGAGCGTGTGCGAACTCAGACC){s<=3}(?P<cell_3>.{7})(?P<discard_3>ATCCACGTGCTTGAGAGGCCAGAGCATTCGAG){s<=3}(?P<cell_4>.{3}).*' \
--stdin=paired-seq/data/SRR8980190_cleaned_R1.fastq.gz \
--stdout=paired-seq/data/SRR8980190_extracted_R1.fastq.gz \
--read2-in=paired-seq/data/SRR8980190_cleaned_R2.fastq.gz \
--read2-out=/dev/null
Explain The Cell Barcode Extraction#
You can check the UMI-tools documentation to see exactly what the command means. Here I explain them briefly:
--extract-method=regex: Use regular expression for the pattern matching.
--bc-pattern2:
Search the pattern in Read 2 using the specified regular expression. If you are familiar with regular expression, this should be relatively straightforward to understand. From 5’ to 5’, it consists of the following part:
(?P<umi_1>.{10}): Capture this 10 bp as UMI.
(?P<cell_1>.{7}): Capture this 7 bp as cell barcode 1.
.{0,3}: this is the 0 - 3 random Ns, we don’t do anything.
(?P<discard_1>GTGGCCGATGTTTCGCATCGGCGTACGACT){s<=3}: We want to discard this linker sequence, with 3 mismatches allowed.
(?P<cell_2>.{7}): Capture this 7 bp as cell barcode 2.
(?P<discard_2>GGATTCGAGGAGCGTGTGCGAACTCAGACC){s<=3}: We want to discard this linker sequence, with 3 mismatches allowed.
(?P<cell_3>.{7}): Capture this 7 bp as cell barcode 3.
(?P<discard_3>ATCCACGTGCTTGAGAGGCCAGAGCATTCGAG){s<=3}: We want to discard this linker sequence, with 3 mismatches allowed.
(?P<cell_4>.{3}): Capture this 3 bp as cell barcode 3.
.*: There may or may not be some leftover sequence which we don’t care.
--stdin: Read 1 file.
--stdout: Read 1 file with extracted cell barcode and UMI information in the header.
--read2-in: Read 2 file.
--read2-out=/dev/null: After getting the cell barcodes and UMI, the Read 2 file does not contain any useful information, so we don’t output it.
Unfortunately, UMI-tools does not output cell barcodes and UMI as separate fastq files at this time of writing (2022-Aug-10). It only extracts the cell barcodes and UMI to the header of the other file, in this case, the header of R1, and we have lost the quality strings at this point. If you look at the first few lines of the output file SRR8980190_extracted_R1.fastq.gz:
@SRR8980190.5_ACCAAGTATAAGGGCCTAAGATCT_AAGCAGAGAC 5/1
AGCCAGAGCATGGAGGAAATGCCTGTCTGCGCCTTCAGATCCCCCTCGTGCGC
+
DDDDDIIIIIIIIIIIIIIIIIIIHIIIHIIIIIIIIIHIHIHIIIIIIIIII
@SRR8980190.6_GCTCGAACCTAGTCATGTGCCTGA_CCGAGTTAAC 6/1
CAGGNTATCCAGTGCAGGTGTTTGAGCAGGTGAATCAGATTGCCATAAGNGCG
+
DDDD#<DEHHIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIHH#<DG
@SRR8980190.7_CATCGATAGGAGGTACCCTTTATC_CTGTGGCTAT 7/1
CTCCACTGTGGCACTTTTTCTCCACCAGACCTCGTGTATATCTCTGTGCTGGG
+
DDDDDIIIHIIIIIIHIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII
We can see the cell barcodes at the header in the 2nd field using _ as the separator. To get this information to a new fastq files, we can use awk and generate fake quality strings. In addition, we don’t really need the UMI for ATAC-seq (maybe it is useful …):
zcat paired-seq/data/SRR8980190_extracted_R1.fastq.gz | \
awk -F '_' '(NR%4==1){print $0 "\n" $2 "\n+\n" "IIIIIIIIIIIIIIIIIIIIIIII"}' | \
gzip > paired-seq/data/SRR8980190_extracted_CB.fastq.gz
After that, we are ready to begin the preprocessing.
Prepare Whitelist#
Cells are barcoded for the first time by either barcoded (3 bp) Tn5 for DNA or oligo-dT primers, followed by three more rounds of ligation. Each round will add 7-bp Ligation Barcode to the molecules. There are 96 different Ligation Barcodes in each round. The same set of 96 Ligation Barcodes are used in each round. Single cells can be identified by the combination of themselves. Here is the information from the Supplementary Table 2 from the Paired-seq paper:
Round 1 barcodes (eight 3-bp Tn5 or oligo-dT barcodes)
Name |
Sequence |
Reverse complement |
|---|---|---|
R01_01 |
ATC |
GAT |
R01_02 |
TGA |
TCA |
R01_03 |
GCT |
AGC |
R01_04 |
CAG |
CTG |
R01_05 |
AGA |
TCT |
R01_06 |
TCT |
AGA |
R01_07 |
GAG |
CTC |
R01_08 |
CTC |
GAG |
Round 2, 3 and 4 barcodes (7 bp)
Name |
Sequence |
Reverse complement |
|---|---|---|
R02/03/04_01 |
AAACCGG |
CCGGTTT |
R02/03/04_02 |
AAACGTC |
GACGTTT |
R02/03/04_03 |
AAAGATG |
CATCTTT |
R02/03/04_04 |
AAATCCA |
TGGATTT |
R02/03/04_05 |
AAATGAG |
CTCATTT |
R02/03/04_06 |
AACACTG |
CAGTGTT |
R02/03/04_07 |
AACGTTT |
AAACGTT |
R02/03/04_08 |
AAGAAGC |
GCTTCTT |
R02/03/04_09 |
AAGCCCT |
AGGGCTT |
R02/03/04_10 |
AAGCTAC |
GTAGCTT |
R02/03/04_11 |
AATCTTG |
CAAGATT |
R02/03/04_12 |
ACAACAC |
GTGTTGT |
R02/03/04_13 |
ACAGTAT |
ATACTGT |
R02/03/04_14 |
ACCAAGT |
ACTTGGT |
R02/03/04_15 |
ACCCTAA |
TTAGGGT |
R02/03/04_16 |
ACCCTTT |
AAAGGGT |
R02/03/04_17 |
ACCTCTC |
GAGAGGT |
R02/03/04_18 |
ACGATTG |
CAATCGT |
R02/03/04_19 |
ACGCAGA |
TCTGCGT |
R02/03/04_20 |
ACGTAAA |
TTTACGT |
R02/03/04_21 |
ACTACCT |
AGGTAGT |
R02/03/04_22 |
ACTCGGT |
ACCGAGT |
R02/03/04_23 |
ACTGTCG |
CGACAGT |
R02/03/04_24 |
ACTTATG |
CATAAGT |
R02/03/04_25 |
AGAAAGG |
CCTTTCT |
R02/03/04_26 |
AGAATCT |
AGATTCT |
R02/03/04_27 |
AGACATA |
TATGTCT |
R02/03/04_28 |
AGAGACC |
GGTCTCT |
R02/03/04_29 |
AGCCCAA |
TTGGGCT |
R02/03/04_30 |
AGCTATT |
AATAGCT |
R02/03/04_31 |
AGGAGGT |
ACCTCCT |
R02/03/04_32 |
AGGGCTT |
AAGCCCT |
R02/03/04_33 |
AGGTGTA |
TACACCT |
R02/03/04_34 |
AGTGCTC |
GAGCACT |
R02/03/04_35 |
AGTGGGA |
TCCCACT |
R02/03/04_36 |
AGTTACG |
CGTAACT |
R02/03/04_37 |
ATAAGGG |
CCCTTAT |
R02/03/04_38 |
ATCATTC |
GAATGAT |
R02/03/04_39 |
ATGGAAC |
GTTCCAT |
R02/03/04_40 |
ATGTGCC |
GGCACAT |
R02/03/04_41 |
ATTCACC |
GGTGAAT |
R02/03/04_42 |
ATTCGAG |
CTCGAAT |
R02/03/04_43 |
CAAGCCT |
AGGCTTG |
R02/03/04_44 |
CACAAGG |
CCTTGTG |
R02/03/04_45 |
CACCTTA |
TAAGGTG |
R02/03/04_46 |
CAGAGTG |
CACTCTG |
R02/03/04_47 |
CAGCGAA |
TTCGCTG |
R02/03/04_48 |
CAGGTCA |
TGACCTG |
R02/03/04_49 |
CATAACT |
AGTTATG |
R02/03/04_50 |
CATATCG |
CGATATG |
R02/03/04_51 |
CATCGAT |
ATCGATG |
R02/03/04_52 |
CATTACA |
TGTAATG |
R02/03/04_53 |
CATTTCC |
GGAAATG |
R02/03/04_54 |
CCAAATG |
CATTTGG |
R02/03/04_55 |
CCACTTG |
CAAGTGG |
R02/03/04_56 |
CCGGATA |
TATCCGG |
R02/03/04_57 |
CCGGTTT |
AAACCGG |
R02/03/04_58 |
CCTAAGA |
TCTTAGG |
R02/03/04_59 |
CCTAGTC |
GACTAGG |
R02/03/04_60 |
CCTGCAA |
TTGCAGG |
R02/03/04_61 |
CGACGTT |
AACGTCG |
R02/03/04_62 |
CGAGTAA |
TTACTCG |
R02/03/04_63 |
CGATTAT |
ATAATCG |
R02/03/04_64 |
CGTAGCA |
TGCTACG |
R02/03/04_65 |
CGTCTGA |
TCAGACG |
R02/03/04_66 |
CTACAGC |
GCTGTAG |
R02/03/04_67 |
CTCAATA |
TATTGAG |
R02/03/04_68 |
CTCGTTG |
CAACGAG |
R02/03/04_69 |
CTCTACG |
CGTAGAG |
R02/03/04_70 |
CTTGGGT |
ACCCAAG |
R02/03/04_71 |
GAAACTC |
GAGTTTC |
R02/03/04_72 |
GACTGTC |
GACAGTC |
R02/03/04_73 |
GATACAG |
CTGTATC |
R02/03/04_74 |
GCGATCA |
TGATCGC |
R02/03/04_75 |
GCGTACT |
AGTACGC |
R02/03/04_76 |
GCTCGAA |
TTCGAGC |
R02/03/04_77 |
GGAAGAA |
TTCTTCC |
R02/03/04_78 |
GGAGATT |
AATCTCC |
R02/03/04_79 |
GGGCTAA |
TTAGCCC |
R02/03/04_80 |
GGGTATG |
CATACCC |
R02/03/04_81 |
GGTAACC |
GGTTACC |
R02/03/04_82 |
GGTAGTG |
CACTACC |
R02/03/04_83 |
GGTGAAA |
TTTCACC |
R02/03/04_84 |
GTAATCG |
CGATTAC |
R02/03/04_85 |
GTATAAG |
CTTATAC |
R02/03/04_86 |
GTCAGAC |
GTCTGAC |
R02/03/04_87 |
GTCCCTT |
AAGGGAC |
R02/03/04_88 |
GTGCCAT |
ATGGCAC |
R02/03/04_89 |
GTGGTCT |
AGACCAC |
R02/03/04_90 |
GTTCTCC |
GGAGAAC |
R02/03/04_91 |
GTTGCTT |
AAGCAAC |
R02/03/04_92 |
TACCCGA |
TCGGGTA |
R02/03/04_93 |
TAGACGA |
TCGTCTA |
R02/03/04_94 |
TAGTCAC |
GTGACTA |
R02/03/04_95 |
TCACATC |
GATGTGA |
R02/03/04_96 |
TCAGCTG |
CAGCTGA |
I have prepared the above two tables as csv files for you, and you can download them:
paired-seq_bc01.csv
paired-seq_bc02-03-04.csv
Since during each ligation round, the same set of Ligation Barcodes (96) are used. Therefore, the whitelist is basically the combination of those 96 barcodes themselves for three times and with those 8 barcodes in the first round: a total of 96 * 96 * 96 * 8 = 7,077,888 barcodes. Since the barcodes are sequenced as Read 2, which uses the top strand as the template, we should use the barcode sequences as they are to construct the whitelist. In addition, the order of the barcodes in Read 2 is Round 4 -> Round 3 -> Round 2 -> Round 1. Therefore, we need to generate the whitelist in this order. Again, if you are confused, check the Paired-seq GitHub page.
# download the barcode files
wget -P paired-seq/data https://teichlab.github.io/scg_lib_structs/data/Paired-seq/paired-seq_bc01.csv \
https://teichlab.github.io/scg_lib_structs/data/Paired-seq/paired-seq_bc02-03-04.csv
# generate whitelist for chromap
for w in $(tail -n +2 paired-seq/data/paired-seq_bc02-03-04.csv | cut -f 2 -d,); do
for x in $(tail -n +2 paired-seq/data/paired-seq_bc02-03-04.csv | cut -f 2 -d,); do
for y in $(tail -n +2 paired-seq/data/paired-seq_bc02-03-04.csv | cut -f 2 -d,); do
for z in $(tail -n +2 paired-seq/data/paired-seq_bc01.csv | cut -f 2 -d,); do
echo "${x}${y}${z}"
done
done
done
done > paired-seq/data/whitelist.txt
# get plain barcode sequence, one per line for starsolo
tail -n +2 paired-seq/data/paired-seq_bc01.csv | \
cut -f 2 -d, > paired-seq/data/round1_bc.txt
tail -n +2 paired-seq/data/paired-seq_bc02-03-04.csv | \
cut -f 2 -d, > paired-seq/data/round234_bc.txt
You will see in the later section why we need those additional files for starsolo. Now we are ready to start the preprocessing.
From FastQ To Count Matrices#
Let’s map the reads to the genome using starsolo for the RNA library and chromap for the ATAC library:
mkdir -p paired-seq/star_outs
mkdir -p paired-seq/chromap_outs
# process the RNA library using starsolo
STAR --runThreadN 4 \
--genomeDir mm10/star_index \
--readFilesCommand zcat \
--outFileNamePrefix paired-seq/star_outs/ \
--readFilesIn paired-seq/data/SRR8980191_cleaned_R1.fastq.gz paired-seq/data/SRR8980191_cleaned_R2.fastq.gz \
--soloType CB_UMI_Complex \
--soloAdapterSequence ATCCACGTGCTTGAGAGGCCAGAGCATTCGTC \
--soloAdapterMismatchesNmax 3 \
--soloCBposition 0_10_0_16 2_-44_2_-38 2_-7_2_-1 2_32_2_34 \
--soloUMIposition 0_0_0_9 \
--soloCBwhitelist paired-seq/data/round234_bc.txt paired-seq/data/round234_bc.txt paired-seq/data/round234_bc.txt paired-seq/data/round1_bc.txt \
--soloCBmatchWLtype 1MM \
--soloCellFilter EmptyDrops_CR \
--outSAMattributes CB UB \
--outSAMtype BAM SortedByCoordinate
# process the ATAC library using chromap
## map and generate the fragment file
chromap -t 4 --preset atac \
-x mm10/chromap_index/genome.index \
-r mm10/mm10.fa \
-1 paired-seq/data/SRR8980190_extracted_R1.fastq.gz \
-b paired-seq/data/SRR8980190_extracted_CB.fastq.gz \
--barcode-whitelist paired-seq/data/whitelist.txt \
-o paired-seq/chromap_outs/fragments.tsv
## compress and index the fragment file
bgzip paired-seq/chromap_outs/fragments.tsv
tabix -s 1 -b 2 -e 3 -p bed paired-seq/chromap_outs/fragments.tsv.gz
After this stage, we are done with the RNA library. The count matrix and other useful information can be found in the star_outs directory. For the ATAC library, two new files fragments.tsv.gz and fragments.tsv.gz.tbi are generated. They will be useful and sometimes required for other programs to perform downstream analysis. There are still some extra work.
Explain star and chromap#
If you understand the Paired-seq experimental procedures described in this GitHub Page and read the manual of the programs, the commands above should be straightforward to understand.
Explain star#
--runThreadN 4
Use 4 cores for the preprocessing. Change accordingly if using more or less cores.
--genomeDir mm10/star_index
Pointing to the directory of the star index. The public data we are analysing is from the cerebral cortex of an adult mouse.
--readFilesCommand zcat
Since the
fastqfiles are in.gzformat, we need thezcatcommand to extract them on the fly.
--outFileNamePrefix paired-seq/star_outs/
We want to keep everything organised. This directs all output files inside the
paired-seq/star_outsdirectory.
--readFilesIn
If you check the manual, we should put two files here. The first file is the reads that come from cDNA, and the second the file should contain cell barcode and UMI. In Paired-seq, cDNA reads come from Read 1. The cell barcode and UMI, together with three linker sequences, come from Read 2. Check the Paired-seq GitHub Page if you are not sure. Multiple input files are supported and they can be listed in a comma-separated manner. In that case, they must be in the same order.
--soloType CB_UMI_Complex
Since Read 2 not only has cell barcodes and UMI, the common linker sequences are also there. The cell barcodes are non-consecutive, separated by the linker sequences. In this case, we have to use the
CB_UMI_Complexoption. Of course, we could also useUMI-toolsto extract the cell barcode and UMI like the ATAC modality, but that’s slow. It is better to use this option. See below.
--soloAdapterSequence ATCCACGTGCTTGAGAGGCCAGAGCATTCGTC
The 0 - 3 random bases in the middle of Read 2 makes the situation complicated, because the absolute positions of the cell barcode and UMI in each read will vary. However, by specifying an adapter sequence, we could use this sequence as an anchor, and tell the program where cell barcodes and UMI are located relatively to the anchor. I choose this linker sequence as the adaptor because it is the longest. However, the other two linker sequences can be used as well. See below.
--soloAdapterMismatchesNmax 3
The number of mismatches are tolerated during the adapter finding. The adapter here is a bit long, so I want a bit relaxed matching, but you may want to try a few different options, like 1 (the default) or 2.
--soloCBposition and --soloUMIposition
These options specify the locations of cell barcode and UMI in the 2nd fastq files we passed to
--readFilesIn. In this case, it is Read 2. Read the STAR manual for more details. I have drawn a picture to help myself decide the exact parameters. There are some freedom here depending on what you are using as anchors. Due to the 3 random bases in the middle, using Read start as anchor will not work for the barcodes in the middle. We need to use the adapter as the anchor, and specify the positions relative to the anchor. See the image:
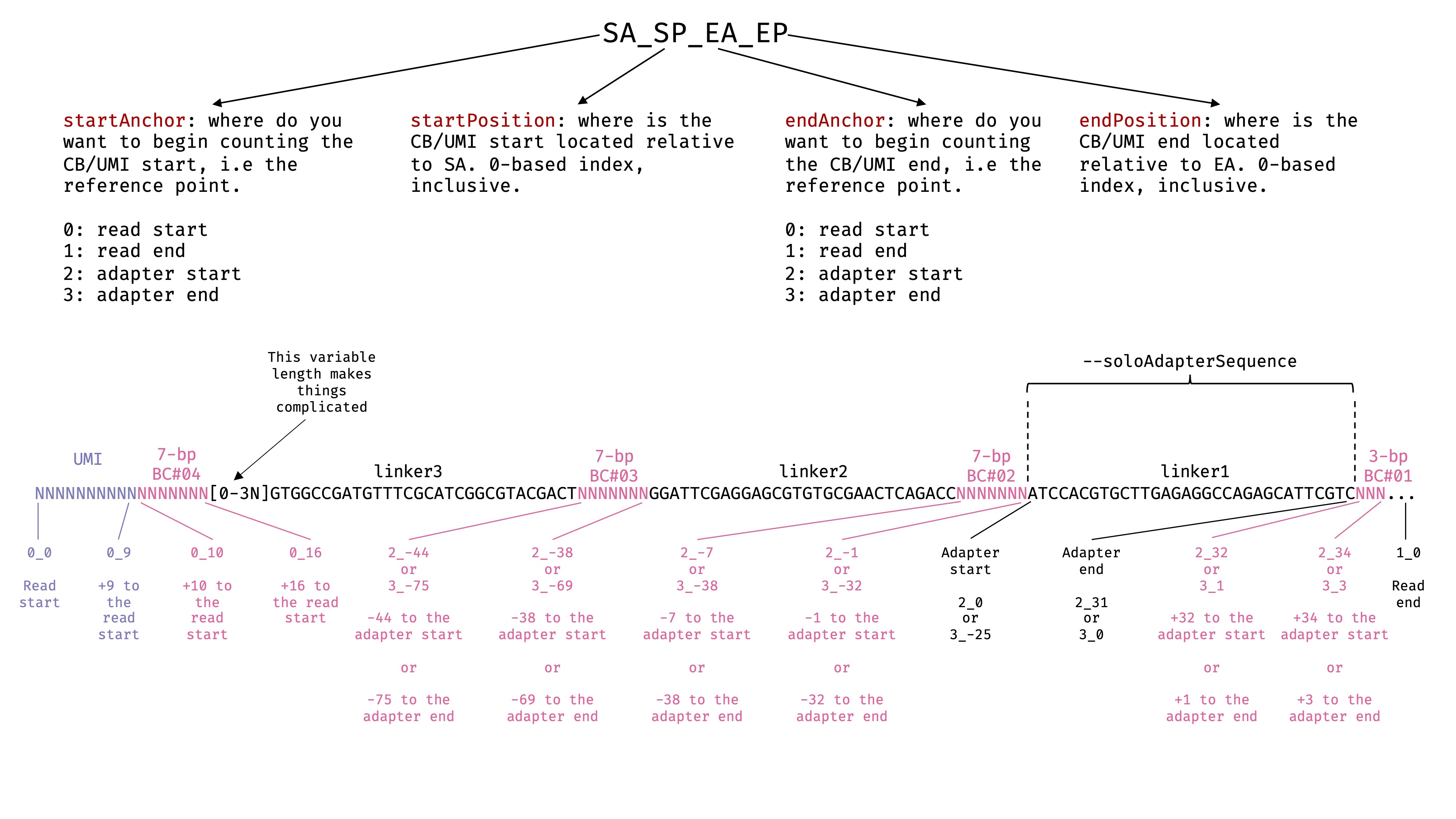
--soloCBwhitelist
Since the real cell barcodes consists of four non-consecutive parts, the whitelist here is the combination of the four sub-lists. We should provide them separately and
starwill take care of the combinations.
--soloCBmatchWLtype 1MM
How stringent we want the cell barcode reads to match the whitelist. The default option (
1MM_Multi) does not work here. We choose this one here for simplicity, but you might want to experimenting different parameters to see what the difference is.
--soloCellFilter EmptyDrops_CR
Experiments are never perfect. Even for barcodes that do not capture the molecules inside the cells, you may still get some reads due to various reasons, such as ambient RNA or DNA and leakage. In general, the number of reads from those cell barcodes should be much smaller, often orders of magnitude smaller, than those barcodes that come from real cells. In order to identify true cells from the background, you can apply different algorithms. Check the
starmanual for more information. We useEmptyDrops_CRwhich is the most frequently used parameter.
--soloStrand Forward
The choice of this parameter depends on where the cDNA reads come from, i.e. the reads from the first file passed to
--readFilesIn. You need to check the experimental protocol. If the cDNA reads are from the same strand as the mRNA (the coding strand), this parameter will beForward(this is the default). If they are from the opposite strand as the mRNA, which is often called the first strand, this parameter will beReverse. In the case of Paired-seq, the cDNA reads are from the Read 1 file. During the experiment, the mRNA molecules are captured by barcoded oligo-dT primer containing UMI and the Read 2 sequence. Therefore, Read 2 consists of cell barcodes and UMI come from the first strand, complementary to the coding strand. Read 1 comes from the coding strand. Therefore, useForwardfor Paired-seq data. ThisForwardparameter is the default, because many protocols generate data like this, but I still specified it here to make it clear. Check the Paired-seq GitHub Page if you are not sure.
--outSAMattributes CB UB
We want the cell barcode and UMI sequences in the
CBandUBattributes of the output, respectively. The information will be very helpful for downstream analysis.
--outSAMtype BAM SortedByCoordinate
We want sorted
BAMfor easy handling by other programs.
Explain chromap#
-t 4
Use 4 cores for the preprocessing. Change accordingly if using more or less cores.
-x mm10/chromap_index/genome.index
The
chromapindex file. The public data we are analysing is from the cerebral cortex of an adult mouse.
-r mm10/mm10.fa
Reference genome sequence in
fastaformat. This is basically the file which you used to create thechromapindex file.
-1, and -b
They are Read 1 (genomic) and cell barcode read, respectively. For ATAC-seq, the sequencing is usually done in pair-end mode. However, the Read 2 in Paired-seq only contains cell barcodes and UMI. Therefore, the ATAC-seq is essentially single-end.
R1is the genomic Read 1 and should be passed to-1; TheCBfile we just prepared contains the cell barcode and should be passed to-b. Multiple input files are supported and they can be listed in a comma-separated manner. In that case, they must be in the same order.
--barcode-whitelist paired-seq/data/whitelist.txt
The plain text file containing all possible valid cell barcodes, one per line. This is the whitelist we just prepared in the previous section. It contains all possible combination of the 96 8-bp barcodes for three times. A total of 96 * 96 * 96 * 8 = 7,077,888 barcodes are in this file.
-o paired-seq/chromap_outs/fragments.tsv
Direct the mapped fragments to a file. The format is described in the 10x Genomics website.
Important
For single-end ATAC-seq, I’m not sure how to interpret the fragment.tsv file, because there is no fragments, just reads in the single-end data. Based on the content and length inside the file, it seems it contains the reads. For the sake of demonstration, I just treat this file as the real fragment file, as if it is from pair-end data.
From ATAC Fragments To Reads#
The fragment file is the following format:
Column number |
Meaning |
|---|---|
1 |
fragment chromosome |
2 |
fragment start |
3 |
fragment end |
4 |
cell barcode |
5 |
Number of read pairs of this fragment |
It is very useful, but we often need the peak-by-cell matrix for the downstream analysis. Therefore, we need to perform a peak calling process to identify open chromatin regions. We need to convert the fragment into reads. For each fragment, we will have two reads, a forward read and a reverse read. The read length is not important, but we could generate a 50-bp read pair for each fragment.
First, we need to create a genome size file, which is a tab delimited file with only two columns. The first column is the chromosome name and the second column is the length of the chromosome in bp:
# we also sort the output by chromosome name
# which will be useful later
faSize -detailed mm10/genome.fa | \
sort -k1,1 > mm10/mm10.chrom.sizes
This is the first 5 lines of mm10/mm10.chrom.sizes:
chr1 195471971
chr10 130694993
chr11 122082543
chr12 120129022
chr13 120421639
Now let’s generate the reads from fragments:
# we use bedClip to remove reads outside the chromosome boundary
# we also remove reads mapped to the mitochondrial genome (chrM)
zcat paired-seq/chromap_outs/fragments.tsv.gz | \
awk 'BEGIN{OFS="\t"}{print $1, $2, $2+50, $4, ".", "+" "\n" $1, $3-50, $3, $4, ".", "-"}' | \
sed '/chrM/d' | \
bedClip stdin mm10/mm10.chrom.sizes stdout | \
sort -k1,1 -k2,2n | \
gzip > paired-seq/chromap_outs/reads.bed.gz
Note we also sort the output reads by sort -k1,1 -k2,2n. In this way, the order of chromosomes in the reads.bed.gz is the same as that in genome.chrom.sizes, which makes downstream processes easier. The output reads.bed.gz are the reads in bed format, with the 4th column holding the cell barcodes.
Peak Calling By MACS2#
Now we can use the newly generated read file for the peak calling using MACS2:
macs2 callpeak -t paired-seq/chromap_outs/reads.bed.gz \
-g mm -f BED -q 0.01 \
--nomodel --shift -100 --extsize 200 \
--keep-dup all \
-B --SPMR \
--outdir paired-seq/chromap_outs \
-n aggregate
Explain MACS2#
The reasons of choosing those specific parameters are a bit more complicated. I have dedicated a post for this a while ago. Please have a look at this post if you are still confused. Note the -g, which is the genome size parameter, is basically the sum of human and mouse. The following output files are particularly useful:
File |
Description |
|---|---|
aggregate_peaks.narrowPeak |
Open chromatin peak locations in the narrowPeak format |
aggregate_peaks.xls |
More information about peaks |
aggregate_treat_pileup.bdg |
Signal tracks. Can be used to generate the bigWig file for visualisation |
Getting The Peak-By-Cell Count Matrix#
Now that we have the peak and reads files, we can compute the number of reads in each peak for each cell. Then we could get the peak-by-cell count matrix. There are different ways of doing this. The following is the method I use.
Find Reads In Peaks Per Cell#
First, we use the aggregate_peaks.narrowPeak file. We only need the first 4 columns (chromosome, start, end, peak ID). You can also remove the peaks that overlap the black list regions. The black list is not available for every species and every build, so I’m not doing it here. We also need to sort the peak to make sure the order of the chromosomes in the peak file is the same as that in the genome.chrom.sizes and reads.bed.gz files. Then we could find the overlap by bedtools. We need to do this in a specific way to get the number of reads in each peak from each cell:
# format and sort peaks
cut -f 1-4 paired-seq/chromap_outs/aggregate_peaks.narrowPeak | \
sort -k1,1 -k2,2n > paired-seq/chromap_outs/aggregate_peaks_sorted.bed
# prepare the overlap
bedtools intersect \
-a paired-seq/chromap_outs/aggregate_peaks_sorted.bed \
-b paired-seq/chromap_outs/reads.bed.gz \
-wo -sorted -g mm10/mm10.chrom.sizes | \
sort -k8,8 | \
bedtools groupby -g 8 -c 4 -o freqdesc | \
gzip > paired-seq/chromap_outs/peak_read_ov.tsv.gz
Explain Finding Reads In Peaks Per Cell#
We start with the command before the first pipe, that is, the intersection part. If you read the manual of the bedtools intersect, it should be straightforward to understand. The -wo option will output the records in both -a file and -b file. Since the reads.bed.gz file has the cell barcode information at the 4th column, we would get an output with both peak and cell information for the overlap. The -sorted -g mm10/mm10.chrom.sizes options make the program use very little memory. Here is an example (top 5 lines) of the output of this part:
chr1 3120021 3120278 aggregate_peak_1 chr1 3120049 3120099 TAGTCACAGCTATTGTAATCGTCT . + 50
chr1 3120021 3120278 aggregate_peak_1 chr1 3120052 3120102 TAGTCACAGCTATTGTAATCGTCT . - 50
chr1 3120021 3120278 aggregate_peak_1 chr1 3120062 3120112 GTGGTCTAGAAAGGGGAAGAATGA . - 50
chr1 3120021 3120278 aggregate_peak_1 chr1 3120063 3120113 ACGATTGATAAGGGAAGAAGCTGA . + 50
chr1 3120021 3120278 aggregate_peak_1 chr1 3120064 3120114 GTGGTCTAGAAAGGGGAAGAATGA . + 50
We see that the 8th column holds the cell barcode and we want to group them using bedtools groupby. Therefore, we need to sort by this column, that is the sort -k8,8. When we group by the 8th column, we are interested in how many times each peak appear per group, so we could gather the information of the peak ID (4th column). That is the -g 8 -c 4 -o freqdesc. The -o freqdesc option returns a value:frequency pair in descending order. Here are some records from peak_read_ov.tsv.gz:
AAACCGGAAACCGGAGCTATTTGA aggregate_peak_19496:6
AAACCGGAAACCGGATAAGGGTGA aggregate_peak_30191:2
AAACCGGAAACCGGCAGAGTGCTC aggregate_peak_65730:12
In a way, that is sort of a count matrix in an awkward format. For example:
The first line means that in cell
AAACCGGAAACCGGAGCTATTTGA, the peakaggregate_peak_19496has 6 counts. All the rest peaks not mentioned here have 0 counts in this cell.The second line means that in cell
AAACCGGAAACCGGATAAGGGTGA, the peakaggregate_peak_30191has 2 counts. All the rest peaks not mentioned here have 0 counts in this cell.
Output The Peak-By-Cell Matrix#
At this stage, we pretty much have all the things needed. Those two files aggregate_peaks_sorted.bed and peak_read_ov.tsv.gz contain all information for a peak-by-cell count matrix. We just need a final touch to make the output in a standard format: a market exchange format (MEX). Since most downstream software takes the input from the 10x Genomics Single Cell ATAC results, we are going to generate the MEX and the associated files similar to the output from 10x Genomics.
Here, I’m using a python script for this purpose. You don’t have to do this. Choose whatever works for you. The point here is to just generate similar files as the peak-barcode matrix described from the 10x Genomics website.
First, let’s make a directory to hold the output files and generate the peaks.bed and barcodes.tsv files, which are easy to do:
# create dirctory
mkdir -p paired-seq/chromap_outs/raw_peak_bc_matrix
# The 10x Genomics peaks.bed is a 3-column bed file, so we do
cut -f 1-3 paired-seq/chromap_outs/aggregate_peaks_sorted.bed > \
paired-seq/chromap_outs/raw_peak_bc_matrix/peaks.bed
# The barcode is basically the first column of the file peak_read_ov.tsv.gz
zcat paired-seq/chromap_outs/peak_read_ov.tsv.gz | \
cut -f 1 > \
paired-seq/chromap_outs/raw_peak_bc_matrix/barcodes.tsv
The slightly more difficult file to generate is matrix.mtx. This is the python script generate_csc_mtx.py for this purpose:
# import helper packages
# most entries in the count matrix is 0, so we are going to use a sparse matrix
# since we need to keep updating the sparse matrix, we use lil_matrix from scipy
import sys
import gzip
from scipy.io import mmwrite
from scipy.sparse import lil_matrix
# the unique peak ID is a good renference
# generate a dictionary with peak_id : index_in_the_file
# sys.argv[1] is the 4-column bed file aggregate_peaks_sorted.bed
peak_idx = {}
with open(sys.argv[1]) as fh:
for i, line in enumerate(fh):
_, _, _, peak_name = line.strip().split('\t')
peak_idx[peak_name] = i
# determine and create the dimension of the output matrix
# that is, to calculate the number of peaks and the number of barcodes
# sys.argv[2] is barcodes.tsv
num_peaks = len(peak_idx.keys())
num_cells = len(open(sys.argv[2]).readlines())
mtx = lil_matrix((num_peaks, num_cells), dtype = int)
# read the information from peak_read_ov.tsv.gz
# update the counts into the mtx object
# sys.argv[3] is peak_read_ov.tsv.gz
with gzip.open(sys.argv[3], 'rt') as fh:
for i, line in enumerate(fh):
col_idx = i # each column is a cell barcode
count_info = line.strip().split('\t')[1]
items = count_info.split(',')
for pn_count in items:
pn, count = pn_count.split(':')
row_idx = peak_idx[pn] # each row is a peak
mtx[row_idx, col_idx] = int(count)
# get a CSC sparse matrix, which is the same as the 10x Genomics matrix.mtx
mtx = mtx.tocsc()
# sys.argv[4] is the path to the output directory
mmwrite(sys.argv[4] + '/matrix.mtx', mtx, field='integer')
Run that script in the terminal:
python generate_csc_mtx.py \
paired-seq/chromap_outs/aggregate_peaks_sorted.bed \
paired-seq/chromap_outs/raw_peak_bc_matrix/barcodes.tsv \
paired-seq/chromap_outs/peak_read_ov.tsv.gz \
paired-seq/chromap_outs/raw_peak_bc_matrix
After that, you should have the matrix.mtx in the paired-seq/chromap_outs/raw_peak_bc_matrix directory.
Cell Calling (Filter Cell Barcodes)#
Experiments are never perfect. Even for droplets that do not contain any cell, you may still get some reads. In general, the number of reads from those droplets should be much smaller, often orders of magnitude smaller, than those droplets with cells. In order to identify true cells from the background, we could use starolo. It is used for scRNA-seq in general, but it does have a cell calling function that takes a directory containing raw mtx and associated files, and return the filtered ones. Since starsolo looks for the following three files in the input directory: matrix.mtx, features.tsv and barcodes.tsv. Those are the output from the 10x Genomics scRNA-seq workflow. In this case, we can use peaks.bed as our features.tsv:
# trick starsolo to use peaks.bed as features.tsv by creating symlink
ln -s peaks.bed paired-seq/chromap_outs/raw_peak_bc_matrix/features.tsv
# filter cells using starsolo
STAR --runMode soloCellFiltering \
paired-seq/chromap_outs/raw_peak_bc_matrix \
paired-seq/chromap_outs/filtered_peak_bc_matrix/ \
--soloCellFilter EmptyDrops_CR
# rename the new feature.tsv to peaks.bed or just create symlink
ln -s features.tsv paired-seq/chromap_outs/filtered_peak_bc_matrix/peaks.bed
If everything goes well, your directory should look the same as the following:
scg_prep_test/paired-seq/
├── chromap_outs
│ ├── aggregate_control_lambda.bdg
│ ├── aggregate_peaks.narrowPeak
│ ├── aggregate_peaks_sorted.bed
│ ├── aggregate_peaks.xls
│ ├── aggregate_summits.bed
│ ├── aggregate_treat_pileup.bdg
│ ├── filtered_peak_bc_matrix
│ │ ├── barcodes.tsv
│ │ ├── features.tsv
│ │ ├── matrix.mtx
│ │ └── peaks.bed -> features.tsv
│ ├── fragments.tsv.gz
│ ├── peak_read_ov.tsv.gz
│ ├── raw_peak_bc_matrix
│ │ ├── barcodes.tsv
│ │ ├── features.tsv -> peaks.bed
│ │ ├── matrix.mtx
│ │ └── peaks.bed
│ └── reads.bed.gz
├── data
│ ├── paired-seq_bc01.csv
│ ├── paired-seq_bc02-03-04.csv
│ ├── round1_bc.txt
│ ├── round234_bc.txt
│ ├── SRR8980190_1.fastq.gz
│ ├── SRR8980190_2.fastq.gz
│ ├── SRR8980190_cleaned_R1.fastq.gz
│ ├── SRR8980190_cleaned_R2.fastq.gz
│ ├── SRR8980190_extracted_CB.fastq.gz
│ ├── SRR8980190_extracted_R1.fastq.gz
│ ├── SRR8980191_1.fastq.gz
│ ├── SRR8980191_2.fastq.gz
│ ├── SRR8980191_cleaned_R1.fastq.gz
│ ├── SRR8980191_cleaned_R2.fastq.gz
│ └── whitelist.txt
└── star_outs
├── Aligned.sortedByCoord.out.bam
├── Log.final.out
├── Log.out
├── Log.progress.out
├── SJ.out.tab
└── Solo.out
├── Barcodes.stats
└── Gene
├── Features.stats
├── filtered
│ ├── barcodes.tsv
│ ├── features.tsv
│ └── matrix.mtx
├── raw
│ ├── barcodes.tsv
│ ├── features.tsv
│ └── matrix.mtx
├── Summary.csv
└── UMIperCellSorted.txt
9 directories, 47 files