SPLiT-seq#
Check this GitHub page to see how SPLiT-seq libraries are generated experimentally. This is a split-pool based combinatorial indexing strategy, where fixed cells are used as the reaction chamber. mRNA molecules are first marked by oligo-dT primer with distinct barcodes (Round1 barcodes) in 48 minibulk reactions in a plate. Then all cells are pooled and randomly distributed into wells of a new 96 well plate where another level of barcode (Round2 barcodes) is added. The same procedure is repeated to add a third level of barcodes (Round3 barcodes). After that, cells are pooled, counted and split into sublibraries. For each sublibrary, an i7 index is added. Single cells can be identified by the combination of the Round1 + Round2 + Round3 + i7 barcodes.
Important
Be aware that there are different versions of SPLiT-seq. They have slightly different adaptor sequences and hence require slightly different parameters for the preprocessing steps. Check the SPLiT-seq GitHub Page and this thread for more details. In this documentation, we are using the Science 2018 version.
For Your Own Experiments#
The read configuration is the same as a standard library:
Order |
Read |
Cycle |
Description |
|---|---|---|---|
1 |
Read 1 |
>50 |
This yields |
2 |
Index 1 (i7) |
6 |
This yields |
3 |
Index 2 (i5) |
Optional |
This yields |
4 |
Read 2 |
94 |
This yields |
The content of Read 2 is like this:
Length |
Sequence (5’ -> 3’) |
|---|---|
34 |
10 bp UMI + 8 bp Round3 barcode + GTGGCCGATGTTTCGCATCGGCGTACGACT + 8 bp Round2 barcode + ATCCACGTGCTTGAGAGGCCAGAGCATTCG + 8 bp Round1 barcode |
You can think of the 8 bp Round1, Round2 and Round3 barcodes as the well barcode for the 1st, 2nd and 3rd plates, respectively. The 6 bp i7 barcode is the index for each sublibrary at the final stage. For a cell, it can go into a well in the 1st plate, then another well in the 2nd plate, then another well in the 3rd plate and finally into a sublibrary. Different cells have very low chance of going through the same combination of wells in the three plates and the final sublibrary. Therefore, if reads have the same combination of well barcodes and sublibrary index (Round1 + Round2 + Round3 + i7), we can safely think they are from the same cell.
If you sequence the library via your core facility or a company, you need to provide the i7 index sequence you used during the sublibrary PCR. Then you will get two fastq files (R1 and R2) per sublibrary. The total file number will depend on how many sublibraries in the final library preparation step.
If you sequence the library on your own, you need to get the fastq files by running bcl2fastq by yourself. In this case it is better to write a SampleSheet.csv with i7 indices for each sublibrary. This will yield the fastq files similar to those from your core facility or the company. Here is an example of the SampleSheet.csv from a NextSeq run with 4 sublibraries using indices from the Science 2018 paper:
[Header],,,,,,,,,,,
IEMFileVersion,5,,,,,,,,,,
Date,17/12/2019,,,,,,,,,,
Workflow,GenerateFASTQ,,,,,,,,,,
Application,NextSeq FASTQ Only,,,,,,,,,,
Instrument Type,NextSeq/MiniSeq,,,,,,,,,,
Assay,AmpliSeq Library PLUS for Illumina,,,,,,,,,,
Index Adapters,AmpliSeq CD Indexes (384),,,,,,,,,,
Chemistry,Amplicon,,,,,,,,,,
,,,,,,,,,,,
[Reads],,,,,,,,,,,
66,,,,,,,,,,,
94,,,,,,,,,,,
,,,,,,,,,,,
[Settings],,,,,,,,,,,
,,,,,,,,,,,
[Data],,,,,,,,,,,
Sample_ID,Sample_Name,Sample_Plate,Sample_Well,Index_Plate,Index_Plate_Well,I7_Index_ID,index,I5_Index_ID,index2,Sample_Project,Description
Sublib01,,,,,,BC_0076,CAGATC,,,,
Sublib02,,,,,,BC_0077,ACTTGA,,,,
Sublib03,,,,,,BC_0078,GATCAG,,,,
Sublib04,,,,,,BC_0079,TAGCTT,,,,
Simply run bcl2fastq like this:
bcl2fastq --no-lane-splitting \
--ignore-missing-positions \
--ignore-missing-controls \
--ignore-missing-filter \
--ignore-missing-bcls \
-r 4 -w 4 -p 4
Then, you will have R1_001.fastq.gz and R2_001.fastq.gz per sublibrary like this:
Sublib01_S1_R1_001.fastq.gz # 66 bp: cDNA reads
Sublib01_S1_R2_001.fastq.gz # 94 bp: UMI, three rounds of barcode, linkers
Sublib02_S2_R1_001.fastq.gz # 66 bp: cDNA reads
Sublib02_S2_R2_001.fastq.gz # 94 bp: UMI, three rounds of barcode, linkers
Sublib03_S3_R1_001.fastq.gz # 66 bp: cDNA reads
Sublib03_S3_R2_001.fastq.gz # 94 bp: UMI, three rounds of barcode, linkers
Sublib04_S5_R1_001.fastq.gz # 66 bp: cDNA reads
Sublib05_S5_R2_001.fastq.gz # 94 bp: UMI, three rounds of barcode, linkers
That’s it. You are ready to go from here using starsolo. You can and should treat each sublibrary as separate experiments, and single cell can be identified by the combination of the Round1 + Round2 + Round3 barcodes in R2. Each sublibrary needs to be processed independently as if they are from different experiments. For example, if you detect barcode AACGTGAT + AACGTGAT + AACGTGAT in both Sublib01 and Sublib02, you should treat them as different cells. Therefore, we need to generate count matrix for each sublibrary separately, and combine them in the downstream analysis.
The advantage of doing this is that we actually divide each experiment into small chunks, and use the exact the same procedures for each chunk independently. In addition, the whitelist will simply be the combination of the Round1 + Round2 + Round3 barcodes for all the analysis.
Public Data#
For the purpose of demonstration, we will use the SPLiT-seq data from the following paper:
Note
Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Gray L, Peeler DJ, Mukherjee S, Chen W, Pun SH, Sellers DL, Tasic B, Seelig G (2018) Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360:eaam8999. https://doi.org/10.1126/science.aam8999
where the authors developed SPLiT-seq for the first time and described the details of the method. The data is in GEO under the accession code GSE110823. You can get all the fastq files directly from this ENA page. There are a few sample, and we are going to use the 150000_CNS_nuclei sample. The sample accession is SAMN08567262. As you can see, there are a total of 14 run accessions. Each run accession represents the data from a sublibrary. This means the authors already demultiplexed the data based on i7 index for us. We could just download each run accession and process them independently. Single cells can be identified by the combination of the Round1 + Round2 + Round3 barcodes.
I’m not going to do all 14 sublibraries. Let’s just use the data SRR6750042 for the demonstration:
# get fastq files
mkdir -p mkdir -p split-seq/data
wget -P split-seq/data -c \
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR675/002/SRR6750042/SRR6750042_1.fastq.gz \
ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR675/002/SRR6750042/SRR6750042_2.fastq.gz
Prepare Whitelist#
The full oligo sequences can be found in the Supplementary Table S12 from the SPLiT-seq paper. As you can see, there are a total of 96 different Round2 barcodes and 96 different Round3 barcodes. For the sublibrary index, they provided 8 different ones (BC_0076 - BC_0083), but you can cerntainly do more. For the Round1 barcodes, it is a bit more complicated. There are 96 of them (Round1_01 - Round1_96). The first 48 are oligo-dT primers and the last 48 are random hexamers. They mix them into 48 different wells:
Round1_01 and Round1_49 are mixed in the same well;
Round1_02 and Round1_50 are mixed in the same well;
Round1_03 and Round1_51 are mixed in the same well;
…
…
…
Round1_48 and Round1_96 are mixed in the same well.
Therefore, we actually have 48 different Round1_barcodes. If you use the oligos provided in the Supplementary Table S12 from the SPLiT-seq paper, you should have a capacity of 48 * 96 * 96 * 8 = 3,538,944 combinations. For the preprocessing, we could treat the different Round1 barcodes as if there are 96 different ones. During the downstream analysis after the preprocessing, we could merge them.
I have collected the index table as follows, and the names of the oligos are directly taken from the paper to be consistent:
Round1 Barcodes (8 bp)
Name |
Sequence |
Reverse complement |
|---|---|---|
Round1_01 |
AACGTGAT |
ATCACGTT |
Round1_02 |
AAACATCG |
CGATGTTT |
Round1_03 |
ATGCCTAA |
TTAGGCAT |
Round1_04 |
AGTGGTCA |
TGACCACT |
Round1_05 |
ACCACTGT |
ACAGTGGT |
Round1_06 |
ACATTGGC |
GCCAATGT |
Round1_07 |
CAGATCTG |
CAGATCTG |
Round1_08 |
CATCAAGT |
ACTTGATG |
Round1_09 |
CGCTGATC |
GATCAGCG |
Round1_10 |
ACAAGCTA |
TAGCTTGT |
Round1_11 |
CTGTAGCC |
GGCTACAG |
Round1_12 |
AGTACAAG |
CTTGTACT |
Round1_13 |
AACAACCA |
TGGTTGTT |
Round1_14 |
AACCGAGA |
TCTCGGTT |
Round1_15 |
AACGCTTA |
TAAGCGTT |
Round1_16 |
AAGACGGA |
TCCGTCTT |
Round1_17 |
AAGGTACA |
TGTACCTT |
Round1_18 |
ACACAGAA |
TTCTGTGT |
Round1_19 |
ACAGCAGA |
TCTGCTGT |
Round1_20 |
ACCTCCAA |
TTGGAGGT |
Round1_21 |
ACGCTCGA |
TCGAGCGT |
Round1_22 |
ACGTATCA |
TGATACGT |
Round1_23 |
ACTATGCA |
TGCATAGT |
Round1_24 |
AGAGTCAA |
TTGACTCT |
Round1_25 |
AGATCGCA |
TGCGATCT |
Round1_26 |
AGCAGGAA |
TTCCTGCT |
Round1_27 |
AGTCACTA |
TAGTGACT |
Round1_28 |
ATCCTGTA |
TACAGGAT |
Round1_29 |
ATTGAGGA |
TCCTCAAT |
Round1_30 |
CAACCACA |
TGTGGTTG |
Round1_31 |
GACTAGTA |
TACTAGTC |
Round1_32 |
CAATGGAA |
TTCCATTG |
Round1_33 |
CACTTCGA |
TCGAAGTG |
Round1_34 |
CAGCGTTA |
TAACGCTG |
Round1_35 |
CATACCAA |
TTGGTATG |
Round1_36 |
CCAGTTCA |
TGAACTGG |
Round1_37 |
CCGAAGTA |
TACTTCGG |
Round1_38 |
CCGTGAGA |
TCTCACGG |
Round1_39 |
CCTCCTGA |
TCAGGAGG |
Round1_40 |
CGAACTTA |
TAAGTTCG |
Round1_41 |
CGACTGGA |
TCCAGTCG |
Round1_42 |
CGCATACA |
TGTATGCG |
Round1_43 |
CTCAATGA |
TCATTGAG |
Round1_44 |
CTGAGCCA |
TGGCTCAG |
Round1_45 |
CTGGCATA |
TATGCCAG |
Round1_46 |
GAATCTGA |
TCAGATTC |
Round1_47 |
CAAGACTA |
TAGTCTTG |
Round1_48 |
GAGCTGAA |
TTCAGCTC |
Round1_49 |
GATAGACA |
TGTCTATC |
Round1_50 |
GCCACATA |
TATGTGGC |
Round1_51 |
GCGAGTAA |
TTACTCGC |
Round1_52 |
GCTAACGA |
TCGTTAGC |
Round1_53 |
GCTCGGTA |
TACCGAGC |
Round1_54 |
GGAGAACA |
TGTTCTCC |
Round1_55 |
GGTGCGAA |
TTCGCACC |
Round1_56 |
GTACGCAA |
TTGCGTAC |
Round1_57 |
GTCGTAGA |
TCTACGAC |
Round1_58 |
GTCTGTCA |
TGACAGAC |
Round1_59 |
GTGTTCTA |
TAGAACAC |
Round1_60 |
TAGGATGA |
TCATCCTA |
Round1_61 |
TATCAGCA |
TGCTGATA |
Round1_62 |
TCCGTCTA |
TAGACGGA |
Round1_63 |
TCTTCACA |
TGTGAAGA |
Round1_64 |
TGAAGAGA |
TCTCTTCA |
Round1_65 |
TGGAACAA |
TTGTTCCA |
Round1_66 |
TGGCTTCA |
TGAAGCCA |
Round1_67 |
TGGTGGTA |
TACCACCA |
Round1_68 |
TTCACGCA |
TGCGTGAA |
Round1_69 |
AACTCACC |
GGTGAGTT |
Round1_70 |
AAGAGATC |
GATCTCTT |
Round1_71 |
AAGGACAC |
GTGTCCTT |
Round1_72 |
AATCCGTC |
GACGGATT |
Round1_73 |
AATGTTGC |
GCAACATT |
Round1_74 |
ACACGACC |
GGTCGTGT |
Round1_75 |
ACAGATTC |
GAATCTGT |
Round1_76 |
AGATGTAC |
GTACATCT |
Round1_77 |
AGCACCTC |
GAGGTGCT |
Round1_78 |
AGCCATGC |
GCATGGCT |
Round1_79 |
AGGCTAAC |
GTTAGCCT |
Round1_80 |
ATAGCGAC |
GTCGCTAT |
Round1_81 |
ATCATTCC |
GGAATGAT |
Round1_82 |
ATTGGCTC |
GAGCCAAT |
Round1_83 |
CAAGGAGC |
GCTCCTTG |
Round1_84 |
CACCTTAC |
GTAAGGTG |
Round1_85 |
CCATCCTC |
GAGGATGG |
Round1_86 |
CCGACAAC |
GTTGTCGG |
Round1_87 |
CCTAATCC |
GGATTAGG |
Round1_88 |
CCTCTATC |
GATAGAGG |
Round1_89 |
CGACACAC |
GTGTGTCG |
Round1_90 |
CGGATTGC |
GCAATCCG |
Round1_91 |
CTAAGGTC |
GACCTTAG |
Round1_92 |
GAACAGGC |
GCCTGTTC |
Round1_93 |
GACAGTGC |
GCACTGTC |
Round1_94 |
GAGTTAGC |
GCTAACTC |
Round1_95 |
GATGAATC |
GATTCATC |
Round1_96 |
GCCAAGAC |
GTCTTGGC |
Round2 Barcodes (8 bp)
Name |
Sequence |
Reverse complement |
|---|---|---|
Round2_01 |
AACGTGAT |
ATCACGTT |
Round2_02 |
AAACATCG |
CGATGTTT |
Round2_03 |
ATGCCTAA |
TTAGGCAT |
Round2_04 |
AGTGGTCA |
TGACCACT |
Round2_05 |
ACCACTGT |
ACAGTGGT |
Round2_06 |
ACATTGGC |
GCCAATGT |
Round2_07 |
CAGATCTG |
CAGATCTG |
Round2_08 |
CATCAAGT |
ACTTGATG |
Round2_09 |
CGCTGATC |
GATCAGCG |
Round2_10 |
ACAAGCTA |
TAGCTTGT |
Round2_11 |
CTGTAGCC |
GGCTACAG |
Round2_12 |
AGTACAAG |
CTTGTACT |
Round2_13 |
AACAACCA |
TGGTTGTT |
Round2_14 |
AACCGAGA |
TCTCGGTT |
Round2_15 |
AACGCTTA |
TAAGCGTT |
Round2_16 |
AAGACGGA |
TCCGTCTT |
Round2_17 |
AAGGTACA |
TGTACCTT |
Round2_18 |
ACACAGAA |
TTCTGTGT |
Round2_19 |
ACAGCAGA |
TCTGCTGT |
Round2_20 |
ACCTCCAA |
TTGGAGGT |
Round2_21 |
ACGCTCGA |
TCGAGCGT |
Round2_22 |
ACGTATCA |
TGATACGT |
Round2_23 |
ACTATGCA |
TGCATAGT |
Round2_24 |
AGAGTCAA |
TTGACTCT |
Round2_25 |
AGATCGCA |
TGCGATCT |
Round2_26 |
AGCAGGAA |
TTCCTGCT |
Round2_27 |
AGTCACTA |
TAGTGACT |
Round2_28 |
ATCCTGTA |
TACAGGAT |
Round2_29 |
ATTGAGGA |
TCCTCAAT |
Round2_30 |
CAACCACA |
TGTGGTTG |
Round2_31 |
GACTAGTA |
TACTAGTC |
Round2_32 |
CAATGGAA |
TTCCATTG |
Round2_33 |
CACTTCGA |
TCGAAGTG |
Round2_34 |
CAGCGTTA |
TAACGCTG |
Round2_35 |
CATACCAA |
TTGGTATG |
Round2_36 |
CCAGTTCA |
TGAACTGG |
Round2_37 |
CCGAAGTA |
TACTTCGG |
Round2_38 |
CCGTGAGA |
TCTCACGG |
Round2_39 |
CCTCCTGA |
TCAGGAGG |
Round2_40 |
CGAACTTA |
TAAGTTCG |
Round2_41 |
CGACTGGA |
TCCAGTCG |
Round2_42 |
CGCATACA |
TGTATGCG |
Round2_43 |
CTCAATGA |
TCATTGAG |
Round2_44 |
CTGAGCCA |
TGGCTCAG |
Round2_45 |
CTGGCATA |
TATGCCAG |
Round2_46 |
GAATCTGA |
TCAGATTC |
Round2_47 |
CAAGACTA |
TAGTCTTG |
Round2_48 |
GAGCTGAA |
TTCAGCTC |
Round2_49 |
GATAGACA |
TGTCTATC |
Round2_50 |
GCCACATA |
TATGTGGC |
Round2_51 |
GCGAGTAA |
TTACTCGC |
Round2_52 |
GCTAACGA |
TCGTTAGC |
Round2_53 |
GCTCGGTA |
TACCGAGC |
Round2_54 |
GGAGAACA |
TGTTCTCC |
Round2_55 |
GGTGCGAA |
TTCGCACC |
Round2_56 |
GTACGCAA |
TTGCGTAC |
Round2_57 |
GTCGTAGA |
TCTACGAC |
Round2_58 |
GTCTGTCA |
TGACAGAC |
Round2_59 |
GTGTTCTA |
TAGAACAC |
Round2_60 |
TAGGATGA |
TCATCCTA |
Round2_61 |
TATCAGCA |
TGCTGATA |
Round2_62 |
TCCGTCTA |
TAGACGGA |
Round2_63 |
TCTTCACA |
TGTGAAGA |
Round2_64 |
TGAAGAGA |
TCTCTTCA |
Round2_65 |
TGGAACAA |
TTGTTCCA |
Round2_66 |
TGGCTTCA |
TGAAGCCA |
Round2_67 |
TGGTGGTA |
TACCACCA |
Round2_68 |
TTCACGCA |
TGCGTGAA |
Round2_69 |
AACTCACC |
GGTGAGTT |
Round2_70 |
AAGAGATC |
GATCTCTT |
Round2_71 |
AAGGACAC |
GTGTCCTT |
Round2_72 |
AATCCGTC |
GACGGATT |
Round2_73 |
AATGTTGC |
GCAACATT |
Round2_74 |
ACACGACC |
GGTCGTGT |
Round2_75 |
ACAGATTC |
GAATCTGT |
Round2_76 |
AGATGTAC |
GTACATCT |
Round2_77 |
AGCACCTC |
GAGGTGCT |
Round2_78 |
AGCCATGC |
GCATGGCT |
Round2_79 |
AGGCTAAC |
GTTAGCCT |
Round2_80 |
ATAGCGAC |
GTCGCTAT |
Round2_81 |
ATCATTCC |
GGAATGAT |
Round2_82 |
ATTGGCTC |
GAGCCAAT |
Round2_83 |
CAAGGAGC |
GCTCCTTG |
Round2_84 |
CACCTTAC |
GTAAGGTG |
Round2_85 |
CCATCCTC |
GAGGATGG |
Round2_86 |
CCGACAAC |
GTTGTCGG |
Round2_87 |
CCTAATCC |
GGATTAGG |
Round2_88 |
CCTCTATC |
GATAGAGG |
Round2_89 |
CGACACAC |
GTGTGTCG |
Round2_90 |
CGGATTGC |
GCAATCCG |
Round2_91 |
CTAAGGTC |
GACCTTAG |
Round2_92 |
GAACAGGC |
GCCTGTTC |
Round2_93 |
GACAGTGC |
GCACTGTC |
Round2_94 |
GAGTTAGC |
GCTAACTC |
Round2_95 |
GATGAATC |
GATTCATC |
Round2_96 |
GCCAAGAC |
GTCTTGGC |
Round3 Barcodes (8 bp)
Name |
Sequence |
Reverse complement |
|---|---|---|
Round3_01 |
AACGTGAT |
ATCACGTT |
Round3_02 |
AAACATCG |
CGATGTTT |
Round3_03 |
ATGCCTAA |
TTAGGCAT |
Round3_04 |
AGTGGTCA |
TGACCACT |
Round3_05 |
ACCACTGT |
ACAGTGGT |
Round3_06 |
ACATTGGC |
GCCAATGT |
Round3_07 |
CAGATCTG |
CAGATCTG |
Round3_08 |
CATCAAGT |
ACTTGATG |
Round3_09 |
CGCTGATC |
GATCAGCG |
Round3_10 |
ACAAGCTA |
TAGCTTGT |
Round3_11 |
CTGTAGCC |
GGCTACAG |
Round3_12 |
AGTACAAG |
CTTGTACT |
Round3_13 |
AACAACCA |
TGGTTGTT |
Round3_14 |
AACCGAGA |
TCTCGGTT |
Round3_15 |
AACGCTTA |
TAAGCGTT |
Round3_16 |
AAGACGGA |
TCCGTCTT |
Round3_17 |
AAGGTACA |
TGTACCTT |
Round3_18 |
ACACAGAA |
TTCTGTGT |
Round3_19 |
ACAGCAGA |
TCTGCTGT |
Round3_20 |
ACCTCCAA |
TTGGAGGT |
Round3_21 |
ACGCTCGA |
TCGAGCGT |
Round3_22 |
ACGTATCA |
TGATACGT |
Round3_23 |
ACTATGCA |
TGCATAGT |
Round3_24 |
AGAGTCAA |
TTGACTCT |
Round3_25 |
AGATCGCA |
TGCGATCT |
Round3_26 |
AGCAGGAA |
TTCCTGCT |
Round3_27 |
AGTCACTA |
TAGTGACT |
Round3_28 |
ATCCTGTA |
TACAGGAT |
Round3_29 |
ATTGAGGA |
TCCTCAAT |
Round3_30 |
CAACCACA |
TGTGGTTG |
Round3_31 |
GACTAGTA |
TACTAGTC |
Round3_32 |
CAATGGAA |
TTCCATTG |
Round3_33 |
CACTTCGA |
TCGAAGTG |
Round3_34 |
CAGCGTTA |
TAACGCTG |
Round3_35 |
CATACCAA |
TTGGTATG |
Round3_36 |
CCAGTTCA |
TGAACTGG |
Round3_37 |
CCGAAGTA |
TACTTCGG |
Round3_38 |
CCGTGAGA |
TCTCACGG |
Round3_39 |
CCTCCTGA |
TCAGGAGG |
Round3_40 |
CGAACTTA |
TAAGTTCG |
Round3_41 |
CGACTGGA |
TCCAGTCG |
Round3_42 |
CGCATACA |
TGTATGCG |
Round3_43 |
CTCAATGA |
TCATTGAG |
Round3_44 |
CTGAGCCA |
TGGCTCAG |
Round3_45 |
CTGGCATA |
TATGCCAG |
Round3_46 |
GAATCTGA |
TCAGATTC |
Round3_47 |
CAAGACTA |
TAGTCTTG |
Round3_48 |
GAGCTGAA |
TTCAGCTC |
Round3_49 |
GATAGACA |
TGTCTATC |
Round3_50 |
GCCACATA |
TATGTGGC |
Round3_51 |
GCGAGTAA |
TTACTCGC |
Round3_52 |
GCTAACGA |
TCGTTAGC |
Round3_53 |
GCTCGGTA |
TACCGAGC |
Round3_54 |
GGAGAACA |
TGTTCTCC |
Round3_55 |
GGTGCGAA |
TTCGCACC |
Round3_56 |
GTACGCAA |
TTGCGTAC |
Round3_57 |
GTCGTAGA |
TCTACGAC |
Round3_58 |
GTCTGTCA |
TGACAGAC |
Round3_59 |
GTGTTCTA |
TAGAACAC |
Round3_60 |
TAGGATGA |
TCATCCTA |
Round3_61 |
TATCAGCA |
TGCTGATA |
Round3_62 |
TCCGTCTA |
TAGACGGA |
Round3_63 |
TCTTCACA |
TGTGAAGA |
Round3_64 |
TGAAGAGA |
TCTCTTCA |
Round3_65 |
TGGAACAA |
TTGTTCCA |
Round3_66 |
TGGCTTCA |
TGAAGCCA |
Round3_67 |
TGGTGGTA |
TACCACCA |
Round3_68 |
TTCACGCA |
TGCGTGAA |
Round3_69 |
AACTCACC |
GGTGAGTT |
Round3_70 |
AAGAGATC |
GATCTCTT |
Round3_71 |
AAGGACAC |
GTGTCCTT |
Round3_72 |
AATCCGTC |
GACGGATT |
Round3_73 |
AATGTTGC |
GCAACATT |
Round3_74 |
ACACGACC |
GGTCGTGT |
Round3_75 |
ACAGATTC |
GAATCTGT |
Round3_76 |
AGATGTAC |
GTACATCT |
Round3_77 |
AGCACCTC |
GAGGTGCT |
Round3_78 |
AGCCATGC |
GCATGGCT |
Round3_79 |
AGGCTAAC |
GTTAGCCT |
Round3_80 |
ATAGCGAC |
GTCGCTAT |
Round3_81 |
ATCATTCC |
GGAATGAT |
Round3_82 |
ATTGGCTC |
GAGCCAAT |
Round3_83 |
CAAGGAGC |
GCTCCTTG |
Round3_84 |
CACCTTAC |
GTAAGGTG |
Round3_85 |
CCATCCTC |
GAGGATGG |
Round3_86 |
CCGACAAC |
GTTGTCGG |
Round3_87 |
CCTAATCC |
GGATTAGG |
Round3_88 |
CCTCTATC |
GATAGAGG |
Round3_89 |
CGACACAC |
GTGTGTCG |
Round3_90 |
CGGATTGC |
GCAATCCG |
Round3_91 |
CTAAGGTC |
GACCTTAG |
Round3_92 |
GAACAGGC |
GCCTGTTC |
Round3_93 |
GACAGTGC |
GCACTGTC |
Round3_94 |
GAGTTAGC |
GCTAACTC |
Round3_95 |
GATGAATC |
GATTCATC |
Round3_96 |
GCCAAGAC |
GTCTTGGC |
I have put those three tables into csv files and you can download them to have a look:
SPLiT-seq_Round1_bc.csv
SPLiT-seq_Round2_bc.csv
SPLiT-seq_Round3_bc.csv
Let’s download them:
wget -P split-seq/data \
https://teichlab.github.io/scg_lib_structs/data/SPLiT-seq/SPLiT-seq_Round1_bc.csv \
https://teichlab.github.io/scg_lib_structs/data/SPLiT-seq/SPLiT-seq_Round2_bc.csv \
https://teichlab.github.io/scg_lib_structs/data/SPLiT-seq/SPLiT-seq_Round3_bc.csv
Now we need to generate the whitelist of those three rounds of barcodes. Those barcodes are sequenced in Read 2 using the top strand as the template. They are in the same direction of the Illumina TruSeq Read 2 sequence. Therefore, we should take their sequences as they are. In addition, if you check the SPLiT-seq GitHub page, you will see that the Round3 barcode is sequenced first, then Round2 barcode and finally Round1 barcode. Therefore, we should pass the whitelist to starsolo in that order. See the next section for more details.
tail -n +2 split-seq/data/SPLiT-seq_Round1_bc.csv | \
cut -f 2 -d, > split-seq/data/round1_whitelist.txt
tail -n +2 split-seq/data/SPLiT-seq_Round2_bc.csv | \
cut -f 2 -d, > split-seq/data/round2_whitelist.txt
tail -n +2 split-seq/data/SPLiT-seq_Round3_bc.csv | \
cut -f 2 -d, > split-seq/data/round3_whitelist.txt
From FastQ To Count Matrix#
We can run starsolo in the following way:
# map and generate the count matrix
STAR --runThreadN 4 \
--genomeDir mm10/star_index \
--readFilesCommand zcat \
--outFileNamePrefix split-seq/star_outs/ \
--readFilesIn split-seq/data/SRR6750042_1.fastq.gz split-seq/data/SRR6750042_2.fastq.gz \
--soloType CB_UMI_Complex \
--soloCBposition 0_10_0_17 0_48_0_55 0_86_0_93 \
--soloUMIposition 0_0_0_9 \
--soloCBwhitelist split-seq/data/round3_whitelist.txt split-seq/data/round2_whitelist.txt split-seq/data/round1_whitelist.txt \
--soloCBmatchWLtype 1MM \
--soloCellFilter EmptyDrops_CR \
--soloStrand Forward \
--outSAMattributes CB UB \
--outSAMtype BAM SortedByCoordinate
Once that is finished, you can do the exact the same thing with all the rest sublibraries. In practice, you can do this via a loop or a pipeline. They can be run independently in parallel.
Explanation#
If you understand the SPLiT-seq experimental procedures described in this GitHub Page, the command above should be straightforward to understand.
--runThreadN 4
Use 4 cores for the preprocessing. Change accordingly if using more or less cores.
--genomeDir mm10/star_index
Pointing to the directory of the star index. The public data from the above paper was produced from mouse brains.
--readFilesCommand zcat
Since the
fastqfiles are in.gzformat, we need thezcatcommand to extract them on the fly.
--outFileNamePrefix split-seq/star_outs/
We want to keep everything organised. This parameter directs all output files into the
split-seq/star_outs/directory.
--readFilesIn
If you check the manual, we should put two files here. The first file is the reads that come from cDNA, and the second file should contain cell barcode and UMI. In SPLiT-seq, cDNA reads come from Read 1, and the cell barcode and UMI come from Read 2. Check the SPLiT-seq GitHub Page if you are not sure.
--soloType CB_UMI_Complex
Since Read 2 not only has cell barcodes and UMI, the common linker sequences are also there. The cell barcodes are non-consecutive, separated by the linker sequences. In this case, we have to use the
CB_UMI_Complexoption. Of course, we could also extract them upfront into a newfastqfile, but that’s slow. It is better to use this option.
--soloCBposition and --soloUMIposition
These options specify the locations of cell barcode and UMI in the 2nd fastq files we passed to
--readFilesIn. In this case, it is Read 2. Read the STAR manual for more details. I have drawn a picture to help myself decide the exact parameters. There are some freedom here depending on what you are using as anchors. in SPLiT-seq, the UMI and cell barcodes are in fixed position in the Read 2. It is relatively straightforward to specify the parameter. See the image:
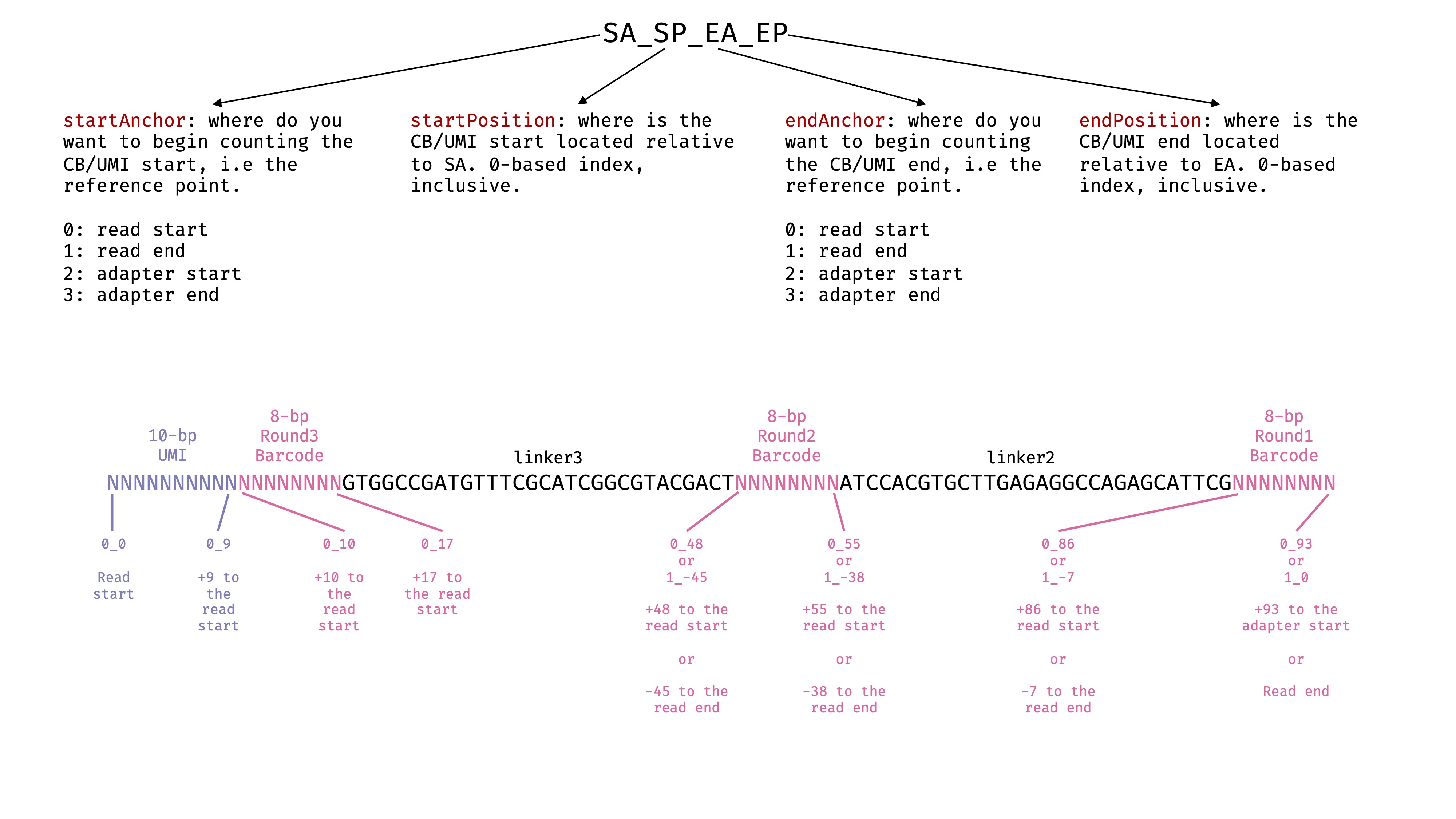
--soloCBwhitelist
Since the real cell barcodes consists of three non-consecutive parts: three rounds of barcodes. The whitelist here is the combination of those three lists. We should provide them separately in the specified order and
starwill take care of the combinations.
--soloCBmatchWLtype 1MM
How stringent we want the cell barcode reads to match the whitelist. The default option (
1MM_Multi) does not work here. We choose this one here for simplicity, but you might want to experimenting different parameters to see what the difference is.
--soloCellFilter EmptyDrops_CR
Experiments are never perfect. Even for barcodes that do not capture the molecules inside the cells, you may still get some reads due to various reasons, such as ambient RNA or DNA and leakage. In general, the number of reads from those cell barcodes should be much smaller, often orders of magnitude smaller, than those barcodes that come from real cells. In order to identify true cells from the background, you can apply different algorithms. Check the
starmanual for more information. We useEmptyDrops_CRwhich is the most frequently used parameter.
--soloStrand Forward
The choice of this parameter depends on where the cDNA reads come from, i.e. the reads from the first file passed to
--readFilesIn. You need to check the experimental protocol. If the cDNA reads are from the same strand as the mRNA (the coding strand), this parameter will beForward(this is the default). If they are from the opposite strand as the mRNA, which is often called the first strand, this parameter will beReverse. In the case of SPLiT-seq, the cDNA reads are from the Read 1 file. During the experiment, the mRNA molecules are captured by barcoded oligo-dT primer containing UMI, and later the Illumina Read 2 sequence will be ligated to this end. Therefore, Read 2 consists of RT barcodes and UMI. They come from the first strand, complementary to the coding strand. Read 1 comes from the coding strand. Therefore, useForwardfor SPLiT-seq data. ThisForwardparameter is the default, because many protocols generate data like this, but I still specified it here to make it clear. Check the SPLiT-seq GitHub Page if you are not sure.
--outSAMattributes CB UB
We want the cell barcode and UMI sequences in the
CBandUBattributes of the output, respectively. The information will be very helpful for downstream analysis.
--outSAMtype BAM SortedByCoordinate
We want sorted
BAMfor easy handling by other programs.
Once that finishes, you could further merge some barcodes based on the information of the Round1 barcodes during the downstream analysis. We are not going to do it here.
If everything goes well, your directory should look the same as the following:
scg_prep_test/split-seq/
├── data
│ ├── round1_whitelist.txt
│ ├── round2_whitelist.txt
│ ├── round3_whitelist.txt
│ ├── SPLiT-seq_Round1_bc.csv
│ ├── SPLiT-seq_Round2_bc.csv
│ ├── SPLiT-seq_Round3_bc.csv
│ ├── SRR6750042_1.fastq.gz
│ └── SRR6750042_2.fastq.gz
└── star_outs
├── Aligned.sortedByCoord.out.bam
├── Log.final.out
├── Log.out
├── Log.progress.out
├── SJ.out.tab
└── Solo.out
├── Barcodes.stats
└── Gene
├── Features.stats
├── filtered
│ ├── barcodes.tsv
│ ├── features.tsv
│ └── matrix.mtx
├── raw
│ ├── barcodes.tsv
│ ├── features.tsv
│ └── matrix.mtx
├── Summary.csv
└── UMIperCellSorted.txt
6 directories, 23 files